
|
like
1 comment
share |
3 min reading time
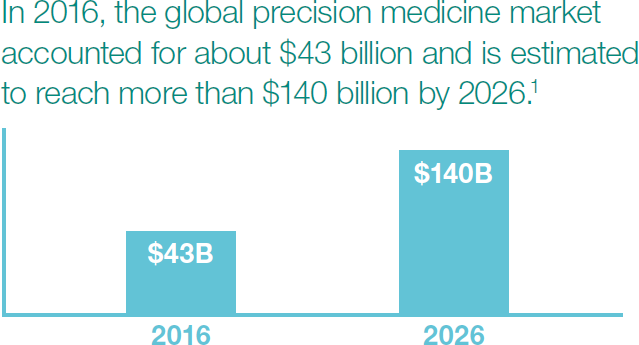
Did you know the market for precision medicine is projected to more than triple by 2026? I’m not surprised by the growth rate given everything we’re reading about personalized medicine. So I got to thinking: How would you regulate something like that?
Taken to extremes, if every device were customized to every single patient, how could the clearance for use be comprehensive enough?
You know this isn’t my personal area of expertise so you may want to download it. I pulled some of these lines for you:
For more information, download the paper, a courtesy of our friends at ICON, plc. FDA webinar in two days.Click here for the FDA content, courtesy of Jon Speer and Greenlight Guru. And you’ll get access to the first FDA in the series where they covered what CfQ is and how FDA is engaging; summary detail on CDRH pilot programs; and, how FDA is rethinking regulations. Members with questionsIf you’ve got the answers, please share with our community. Tautvydas Kazlauskas has a question on how to classify a fibrin-glue like medical device. Antonia Trevisan asks, “Are custom-made devices exempt from CE marking?” And I could really use help on Irina’s question. So far no one has answered, “How to estimate safety BSE requirements?” A whole new worldAs you can see, with our new home here in WordPress, I can add images and video each week. But even better, I have enough room to express myself and to highlight questions and contributions from group members like you! So here’s your chance to ask a question and get a crowd-sourced answer from our members. Just go to https://medicaldevicesgroup.net/question/add/ to ask it. The page will look like this and there’s even a way for you to get notified with each comment (just like the very old days)::
I look forward to your participation! Thank you for being part of our Medical Devices Group community.Make it a great week.
Joe Hage P.S. Meet me at ConX (Sep 17, South Carolina), the MedTech Regulatory Awards at RAPS (Oct 2, Vancouver, BC), and our group’s 10x for Engineers (Oct 10-12, San Diego) – especially that last one! 😊 Marked as spam
Asked on August 13, 2018 11:20 pm
916 views
|


Meet your next client here. Join our medical devices group community.

Private answer
John Abbott
Well… this so-called “precision medicine” is already here and has been, to some degree, for many years. I suspect that most products will be treated as class three requiring a PMA-style review and approval although I can see where some might be down classified through de novo. The difference is that we are talking about a PROCESS as opposed to some piece of hardware or other physical thing (like a drug, for example). In the end it will all come down to a proper risk-management analysis where you have to 1) identify all possible hazards, evaluate their risks and reduce those risks to an acceptable level and 2) be able to demonstrate to the regulator that you did a complete and accurate review and that the resulting product (whether device, drug or process) is safe and effective. The regulatory process is not just some rote checklist or form. Rather it is a requirement on the manufacturer to ensure that their products are safe and effective. I don’t see anything in the existing regulatory process that would not be adequate for precision medicine. And if they don’t already exist, expect the regulatory agencies (like the FDA) to publish guidance documents for how such precision medicine devices/processes will be reviewed and approved. Marked as spam
|
We still use LinkedIn to access our site because it’s the only way to “pull in” your LinkedIn photo, name, and hyperlink to your profile page, all vital in building your professional network. When you log in using LinkedIn, you are giving LinkedIn your password, not me. I never see nor store your LinkedIn credentials.
